bioinfo-statistics
250828-250911 생물정보학 논문 스크리닝 본문

Abstract
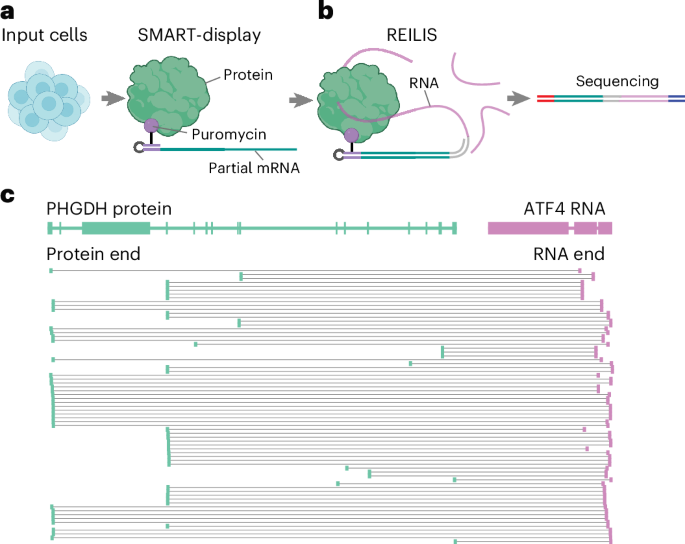
RNA-protein interactions critically regulate gene expression and cellular processes, yet their comprehensive mapping remains challenging due to their structural diversity. We introduce PRIM-seq (protein-RNA interaction mapping by sequencing), a method for concurrent de novo identification of RNA-binding proteins and their associated RNAs. PRIM-seq generates unique chimeric DNA sequences by proximity ligation of RNAs with protein-linked DNA barcodes, which are subsequently decoded through sequencing. We apply PRIM-seq to two human cell lines and construct a human RNA-protein association network (HuRPA), encompassing >350,000 associations involving ~7,000 RNAs and ~11,000 proteins, including 2,610 proteins that each interact with at least 10 distinct RNAs. We experimentally validate the tumorigenesis-associated lincRNA LINC00339, the RNA with the highest number of protein associations in HuRPA, as a protein-associated RNA. We further validate the RNA-associating abilities of chromatin-conformation regulators SMC1A, SMC3 and RAD21, as well as the metabolic enzyme PHGDH. PRIM-seq enables systematic discovery and prioritization of RNA-binding proteins and their targets without gene- or protein-specific reagents.
Main

The problem
- Selecting the optimal molecular assay, or assays, for a particular patient can be challenging for various reasons,
- Gene fusions are more than twice as prevalent in pediatric cancers as in adult cancers1 and are most readily detected by RNA assays
- Here we sought to determine the utility of performing RNA sequencing (RNA-seq) as a standalone up-front test for a targeted panel of cancer-relevant genes in our accredited clinical molecular diagnostics laboratory.
The solution
We assessed RNA-seq results from 2,200 prospectively tested tumor specimens. To test the suitability of RNA-seq for identifying single-nucleotide variants (SNVs), we benchmarked its performance against DNA sequencing (DNA-seq) in samples for which both had been performed on the same sample3.
Although RNA-seq is known to be more sensitive than DNA-seq in identifying gene fusions, we found (in contrast to previous reports4) that RNA-seq was as sensitive as DNA-seq in finding relevant oncogenic SNVs. Moreover, relative to DNA-seq, RNA-seq provided additional information for biological and clinical interpretation, including whether a mutated gene was expressed (and was, therefore, likely to affect function), and whether a splice-site mutation had a functional effect on the expressed transcript (Fig. 1a).
The implications
- A major challenge in molecular pathology is selection of the optimal assay for a particular tumor. Our findings suggest that RNA-seq can function as a single precision test to identify both gene fusions and SNVs in a broad range of cancers.
- Although RNA-seq can robustly identify mutations that are expressed, pathogenic variants that result in the loss of expression (for example, by nonsense-mediated decay) might not be detected by this assay.
- In addition, RNA-seq cannot identify copy-number changes, which are frequently important to consider in diagnostic workflows. Thus, first-line use of RNA-seq cannot always eliminate the need to perform further genetic tests in cancer diagnosis.

Abstract
To understand shared and ancestry-specific genetic control of brain protein expression and its ramifications for disease, we mapped protein quantitative trait loci (pQTLs) in 1,362 brain proteomes from African American, Hispanic/Latin American and non-Hispanic white donors. Among the pQTLs that multiancestry fine-mapping MESuSiE confidently assigned as putative causal pQTLs in a specific population, most were shared across the three studied populations and are referred to as multiancestry causal pQTLs. These multiancestry causal pQTLs were enriched for exonic and promoter regions. To investigate their effects on disease, we modeled the 858 multiancestry causal pQTLs as instrumental variables using Mendelian randomization and genome-wide association study results for neurologic and psychiatric conditions (21 traits in participants with European ancestry, 10 in those with African ancestry and 4 in Hispanic participants). We identified 119 multiancestry pQTL–protein pairs consistent with a causal role in these conditions. Remarkably, 29% of the multiancestry pQTLs in these pairs were coding variants. These results lay an important foundation for the creation of new molecular models of neurologic and psychiatric conditions that are likely to be relevant to individuals across different genetic ancestries.
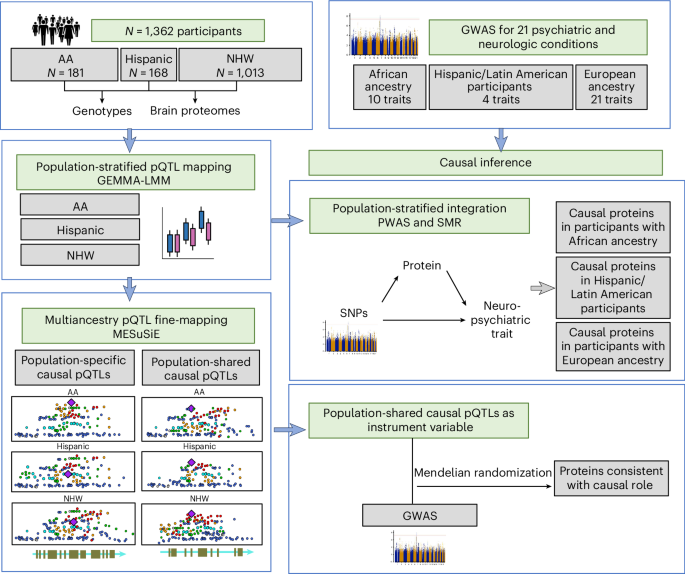
이 논문은 multiancestry brain pQTL(단백질 발현 관련 유전 변이) 분석을 통해 ancestry 간 차이점과 공통점을 체계적으로 살펴봤습니다. 주요 요약은 다음과 같습니다:
1. 공통점
- 대부분의 causal pQTL이 ancestry 간 공유됨
- MESuSiE를 이용한 multiancestry fine-mapping 결과, 약 76% (category.PIP > 0.5) 의 causal pQTL이 세 집단(African American, Hispanic/Latin American, non-Hispanic white, NHW) 모두에서 공유됨
-
- 더 엄격한 기준(category.PIP > 0.9)에서는 99.5%가 공유되어, 거의 모든 high-confidence causal pQTL이 세 집단에서 일관되게 나타났음
-
- 이 공유된 pQTL들은 기능적으로 중요한 영역(UTR, nonsynonymous coding sites 등)에 특히 많이 위치
- 효과 크기의 일관성
- ancestry별 pQTL 효과 크기(beta 값)의 상관관계가 매우 높아(≥0.95, P<2.2×10⁻¹⁶), 세 집단 간 효과 방향과 크기가 거의 동일함
-
- replication rate (π1)도 세 집단 모두 >99%였음
- 질환 연관성의 보편성
- 119개의 multiancestry causal pQTL–protein–trait triad를 규명했으며, 이는 다양한 뇌질환(예: 알츠하이머병, 조현병, 우울증 등)에 세 집단 모두 적용 가능할 가능성이 높음
2. 차이점
- 인구집단 특이적 pQTL 존재
- 일부 pQTL은 특정 집단에서만 나타났음.
-
- 그러나 이러한 사례는 소수였으며, 대부분은 shared.
- 샘플 크기와 GWAS 파워 차이
- NHW 집단(N=1,013)이 AA(N=181), Hispanic(N=168)보다 훨씬 커서, 훨씬 많은 pQTL과 causal proteins을 확인할 수 있었음.
- 예를 들어, NHW에서는 알츠하이머병 관련 64개 causal proteins을 찾았지만, Hispanic 집단에서는 유의한 causal protein이 없음
- GWAS 통합 결과의 차이
- AA 집단에서는 파킨슨병(GBA1)과 알코올 사용장애(METAP1, ADH5)와 같은 일부 인과 단백질만 확인됨.
- Hispanic 집단에서는 GWAS 샘플 수 부족으로 causal protein이 전혀 검출되지 않음
3. 해석
- 연구팀은 거의 모든 고신뢰 causal pQTL이 ancestry 간에 공유됨을 보여줌으로써,
- 현재까지의 차이는 실제 유전적 효과 차이보다는 allele frequency, LD 구조, 샘플 수 차이에서 비롯되었을 가능성이 크다고 결론 내렸습니다
- 동시에, 소수의 ancestry-specific pQTL(특히 AA 집단에서)이 존재하므로, 향후 non-European 집단의 GWAS와 프로테옴 샘플 수 확장이 중요하다고 강조했습니다
👉 정리하면:
이 논문은 대부분의 brain causal pQTL은 ancestry 간 공유되지만, 일부 population-specific 신호가 존재함을 보여줬습니다. 다만 현재 관찰되는 차이의 상당 부분은 샘플 크기와 통계적 파워 차이 때문일 가능성이 크며, 향후 아프리카계·히스패닉 집단 데이터 확대가 필요하다고 했습니다.

Abstract
Gene expression is modulated jointly by transcriptional regulation and messenger RNA stability, yet the latter is often overlooked in studies on genetic variants. Here, leveraging metabolic labeling data (Bru/BruChase-seq) and a new computational pipeline, RNAtracker, we categorize genes as allele-specific RNA stability (asRS) or allele-specific RNA transcription events. We identify more than 5,000 asRS variants among 665 genes across a panel of 11 human cell lines. These variants directly overlap conserved microRNA target regions and allele-specific RNA-binding protein sites, illuminating mechanisms through which stability is mediated. Furthermore, we identified causal asRS variants using a massively parallel screen (MapUTR) for variants that affect post-transcriptional mRNA abundance, as well as through CRISPR prime editing approaches. Notably, asRS genes were enriched significantly among a multitude of immune-related pathways and contribute to the risk of several immune system diseases. This work highlights RNA stability as a critical, yet understudied mechanism linking genetic variation and disease.

Abstract
The spatial resolution of omics analyses is fundamental to understanding tissue biology1,2,3. The capacity to spatially profile DNA methylation, which is a canonical epigenetic mark extensively implicated in transcriptional regulation4,5, is lacking. Here we introduce a method for whole-genome spatial co-profiling of DNA methylation and the transcriptome of the same tissue section at near single-cell resolution. Applying this technology to mouse embryogenesis and the postnatal mouse brain resulted in rich DNA–RNA bimodal tissue maps. These maps revealed the spatial context of known methylation biology and its interplay with gene expression. The concordance and distinction in spatial patterns of the two modalities highlighted a synergistic molecular definition of cell identity in spatial programming of mammalian development and brain function. By integrating spatial maps of mouse embryos at two different developmental stages, we reconstructed the dynamics that underlie mammalian embryogenesis for both the epigenome and transcriptome, revealing details of sequence-, cell-type- and region-specific methylation-mediated transcriptional regulation. This method extends the scope of spatial omics to include DNA cytosine methylation, enabling a more comprehensive understanding of tissue biology across development and disease.

Abstract
Over time, cells in the brain and in the body accumulate damage, which contributes to the ageing process1. In the human brain, the prefrontal cortex undergoes age-related changes that can affect cognitive functioning later in life2. Here, using single-nucleus RNA sequencing (snRNA-seq), single-cell whole-genome sequencing (scWGS) and spatial transcriptomics, we identify gene-expression and genomic changes in the human prefrontal cortex across lifespan, from infancy to centenarian. snRNA-seq identified infant-specific cell clusters enriched for the expression of neurodevelopmental genes, as well as an age-associated common downregulation of cell-essential homeostatic genes that function in ribosomes, transport and metabolism across cell types. Conversely, the expression of neuron-specific genes generally remains stable throughout life. These findings were validated with spatial transcriptomics. scWGS identified two age-associated mutational signatures that correlate with gene transcription and gene repression, respectively, and revealed gene length- and expression-level-dependent rates of somatic mutation in neurons that correlate with the transcriptomic landscape of the aged human brain. Our results provide insight into crucial aspects of human brain development and ageing, and shed light on transcriptomic and genomic dynamics.

Abstract
Coagulation factor XII has been identified as a potential drug target that could prevent thrombosis without increasing the risk of bleeding. However, human data to support the development of factor XII-directed therapeutics are lacking. To assess the role of factor XII in venous thromboembolism, we examine genetic variation in the coding region of the F12 locus across 703,745 participants in the UK Biobank and NIH All of Us biorepositories. We find that heterozygous carriers of nonsense, frameshift, and essential splice site variants in F12 are protected against venous thromboembolism without an increased risk of bleeding or infection. We also show that F12 variant carriers generally experience a quantitative (type I) defect in circulating factor XII levels, though a subset of participants was also identified with possible qualitative (type II) deficiency. In vitro plasma-based thrombin generation is reduced at factor XII concentrations reflective of those seen in F12 variant carriers. We also show that F12 heterozygous mice are protected against venous thromboembolism and display an intermediate phenotype between wild-type and F12-null animals. We conclude that heterozygous loss of F12 represents a haploinsufficient state characterized by protection against venous thromboembolism and that therapeutically inhibiting factor XII is likely to be safe and effective.

Abstract
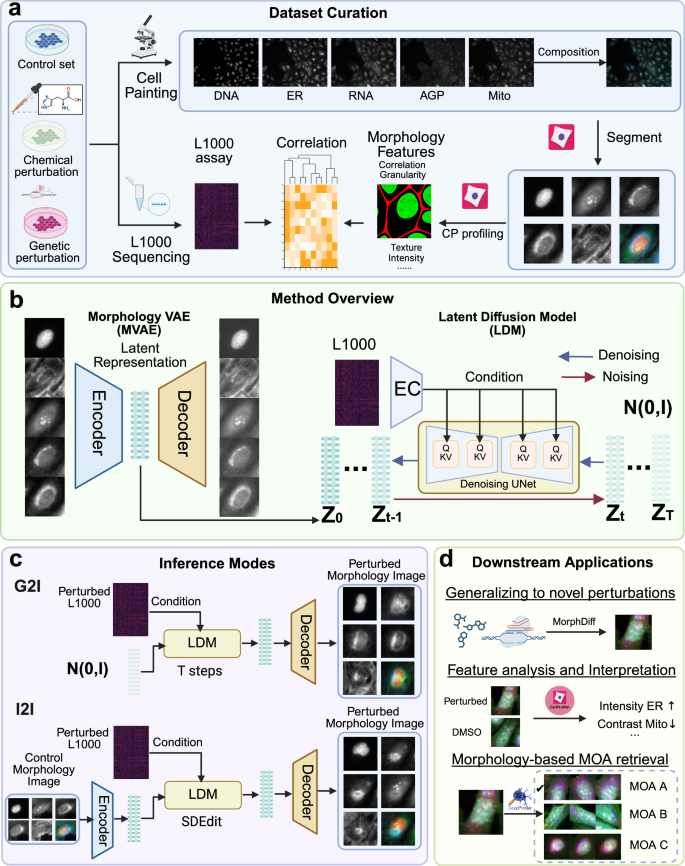
Investigating cell morphology changes after perturbations using high-throughput image-based profiling is increasingly important for phenotypic drug discovery, including predicting mechanisms of action (MOA) and compound bioactivity. The vast space of chemical and genetic perturbations makes it impractical to explore all possibilities using conventional methods. Here we propose MorphDiff, a transcriptome-guided latent diffusion model that simulates high-fidelity cell morphological responses to perturbations. We demonstrate MorphDiff’s effectiveness on three large-scale datasets, including two drug perturbation and one genetic perturbation dataset, covering thousands of perturbations. Extensive benchmarking shows MorphDiff accurately predicts cell morphological changes under unseen perturbations. Additionally, MorphDiff enhances MOA retrieval, achieving an accuracy comparable to ground-truth morphology and outperforming baseline methods by 16.9% and 8.0%, respectively. This work highlights MorphDiff’s potential to accelerate phenotypic screening and improve MOA identification, making it a powerful tool in drug discovery.

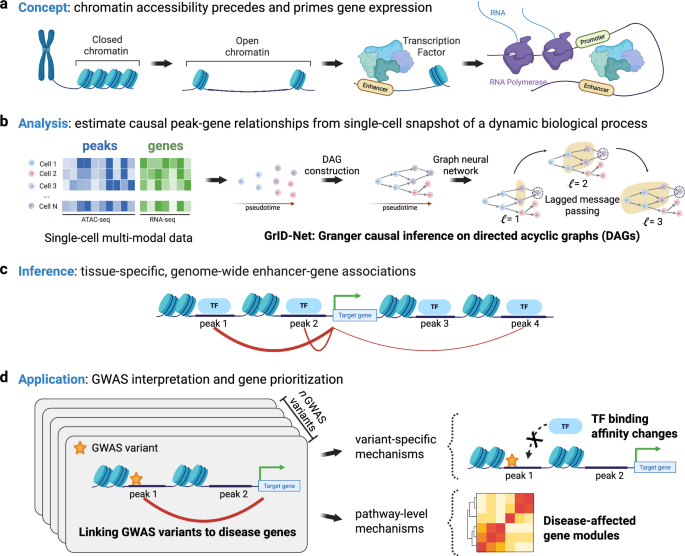
Abstract
Genome-wide association studies (GWAS) identify numerous disease-linked genetic variants at noncoding genomic loci, yet therapeutic progress is hampered by the challenge of deciphering the regulatory roles of these loci in tissue-specific contexts. Single-cell multimodal assays that simultaneously profile chromatin accessibility and gene expression could predict tissue-specific causal links between noncoding loci and the genes they affect. However, current computational strategies either neglect the causal relationship between chromatin accessibility and transcription or lack variant-level precision, aggregating data across genomic ranges due to data sparsity. To address this, we introduce GrID-Net, a graph neural network approach that generalizes Granger causal inference to detect new causal locus–gene associations in graph-structured systems such as single-cell trajectories. Inspired by the principles of optical parallax, which reveals object depth from static snapshots, we hypothesize that causal mechanisms could be inferred from static single-cell snapshots by exploiting the time lag between epigenetic and transcriptional cell states, a concept we term “cell-state parallax.” Applying GrID-Net to schizophrenia (SCZ) genetic variants, we increase variant coverage by 36% and uncovered noncoding mechanisms that dysregulate 132 genes, including key potassium transporters such as KCNG2 and SLC12A6. Furthermore, we discover evidence for the prominent role of neural transcription-factor binding disruptions in SCZ etiology. Our work not only provides a strategy for elucidating the tissue-specific impact of noncoding variants but also underscores the breakthrough potential of cell-state parallax in single-cell multiomics for discovering tissue-specific gene regulatory mechanisms.