

bioinfo-statistics
25.02.08 Nat Med, Artificial intelligence in drug development (2025) 본문
25.02.08 Nat Med, Artificial intelligence in drug development (2025)
spnz3 2025. 2. 8. 17:45논문 링크
https://www.nature.com/articles/s41591-024-03434-4

목차
A. AI-driven drug discovery
1. Target identification
2. Virtual screening
3. De novo design
4. ADMET (absorption, distribution, metabolism, excretion and toxicity)
5. Synthesis planning and automating synthesis and drug discovery
B. AI in clinical trials and real-world practice
1. Biomarker discovery
2. Predicting pharmacometrics properties
3. Drug repurposing
4. Improving trial efficiency and predicting outcomes
C. Challenges
D. Future directions
A. AI-driven drug discovery
1. Target identification
- Identification of small-molecule targets, such as proteins or nucleic acids
- Traditional methods: affinity pull-down and whole-genome knockdown screening
- NLP techniques (such as word2vec embeddings) to map gene functions into high-dimensional space, enhancing the sensitivity of target identification despite the sparsity of gene function overlap26.
- Nonetheless, integrating multi-omics data efficiently and ensuring the interpretability of AI models are challenging tasks. Graph deep learning technology addresses these by merging graph structures with deep learning, focusing on graph nodes related to key features (for example, atom type, charge) to effectively identify candidate targets. A recent study successfully developed an interpretable framework using multi-omics network graphs with graph attention mechanisms to predict cancer genes effectively32.
- Furthermore, integrating multi-omics data with scientific and medical literature into knowledge graphs allows AI to discern relationships between genes and disease pathways33,34,35. Biomedical LLMs, when deeply integrated with biological networks or knowledge graph functions, provide efficient and precise methods for linking diseases, genes and biological processes36. For instance, the PandaOmics platform (https://pharma.ai/pandaomics/) ... However, the potential publication biases in the literature suggest a need for supplementary methods to ensure the identification of novel and relevant targets.
- Real-world data, such as medical records, self-reports, electronic health records (EHRs) and insurance claims, However, real-world data often contain unstructured text, lack standardization and may include biases, limiting their application in this context. ... Nonetheless, recent studies have shown that despite these issues, noisy real-world data can train effective models38 .....
- Enhancing model generalizability across diverse populations remains a major challenge, especially for diseases with low labeling or prevalence rates40.
2. Virtual screening
AI-based receptor–ligand docking models
Notably, recent receptor–ligand co-folding networks based on AlphaFold2 and RosettaFold
However, deep learning-based binding pose prediction models have yet to outperform physics-based methods. Additionally, predicting precise receptor–ligand interaction remains a challenge.
When target structures are absent or incomplete, the direct application of docking-based virtual screening is impractical. As an alternative, AI techniques may be used in sequence-based prediction methods65. However, such methods often struggle to capture the complexity of three-dimensional protein–ligand interactions...
Phenotype-based virtual screening (<- 이거는 target identification에 해당하는 것 아닌가?)
While targeted drug development is effective for defined targets, many diseases lack such targets. Phenotype-based virtual screening is thus crucial for diseases with undefined targets (for example, rare diseases) and broadly phenotypic diseases (for example, aging)66,67,68,69,70,71,72. A recent study used nuclear morphology and machine learning to identify compounds inducing senescence in cancer cells73; similar strategies are also promising for antibiotic discovery68. However, such models often depend on case-specific phenotypic data and struggle with generalization. Furthermore, AI-based activity prediction solely relying on ligand chemical structures faces challenges like data sparsity and imbalance, and activity cliffs...
Challenges
- Current virtual screening models generally focus on specific tasks such as scoring76, pose optimization48 or screening77, emphasizing the need to develop universal models capable of handling multiple tasks78.
- Furthermore, the exponential growth of commercial compound collections to billions makes comprehensive screening computationally infeasible81. Meanwhile, the available molecular libraries cover only a small portion of the druggable chemical space, which continues to expand—creating both opportunities and challenges in navigating and screening for bioactive molecules.
- In response to these challenges, techniques like active learning and Bayesian optimization82 are effective methods for addressing the chemical space search problem
- The integration of quantum mechanics with AI offers new tools for chemical space exploration83, while molecular dynamics simulations add depth to protein–ligand interactions, addressing issues of binding affinity and selectivity to improve model accuracy84. Simultaneously, by generating custom virtual libraries for specific targets or compound types, deep generative models substantially narrow search spaces and enhance screening efficiency85,86,87.
3. De novo design
Successfully applied in developing small-molecule inhibitors87, PROTACs88, peptides89,90,91 and functional proteins92,93 that are validated through wet-lab experiments.
Chemical language models convert molecular generation tasks into sequence generation85,87,94,95,96,97 such as SMILES string (‘simplified molecular input line entry system’, a notation system that represents a chemical structure in a linear text format).
Graph-based models represent molecules as graphs, generating structures using autoregressive or non-autoregressive strategies100,101,102,103. Autoregressive approaches construct molecules atom by atom, which can lead to chemically implausible intermediates and introduce bias101,102. In contrast, non-autoregressive methods generate entire molecular graphs at once but need extra steps to ensure the graph’s validity, as these models’ limited perception of molecular topological structures can induce flawed structures104.

4. ADMET (absorption, distribution, metabolism, excretion and toxicity)
ADMET plays a critical role in determining drug efficacy and safety.
For instance, Bayer’s in silico ADMET platform uses machine learning techniques such as random forest and support vector machines, using descriptors like circular extended connectivity fingerprints to ensure accuracy and relevance120. Over the past decades, various descriptors for ADMET predictions have been developed121,122,123,124. However, feature engineering involved in these feature-based methods remains complex and limits generality and flexibility.
Deep learning now drives ADMET prediction, automatically extracting meaningful features from simple input data. Various neural network architectures, including transformers (designed to effectively handle sequential data)125,126,127, convolutional neural networks (a type of deep learning model commonly used for image and video recognition tasks)128 and, more recently, graph neural networks (deep learning models for processing graph-structured data, such as molecular structures)78,129, excel in modeling molecular properties from formats such as SMILES strings and molecular graphs.... For predictions involving properties like toxicity, the performance of representations generated by these models might saturate before training progresses, showing limited improvement after training.
Challenge
- Scarce labeled data in ADMET predictions
- While large transformer-based models show promise in other fields, their use in ADMET prediction remains underexplored. A recent study130 indicates that although SMILES language does not encode molecular topology directly, carefully designed self-supervised training with contextual transformers equipped with linear attention mechanisms can effectively learn implicit structure–property relationships, bolstering confidence in applying large-scale self-supervised models for ADMET predictions.
- Molecular representation is critical for AI performance. High-dimensional representations typically provide richer information than low-dimensional ones. Multimodal ADMET models using multiple representations simultaneously holds promise, although the optimal combination of data types is still unresolved.
- Interpretability Attention mechanisms, integrating chemical knowledge136 can further enhance interpretability...
5. Synthesis planning and automating synthesis and drug discovery
Chemical synthesis, one of the bottlenecks in small-molecule drug discovery, is a highly technical and extremely laborious task. Computer-aided synthesis planning (CASP) and automatic synthesis of organic compounds can help alleviate the burden of repetitive laborious tasks for chemists, enabling them to engage in more innovative works137,138.
...(화학 쪽은 잘 몰라서 생략)...
Following the planning and synthesis of new drug compounds, AI technology facilities the in vivo validation of the mechanism of action (MOA) of new drugs. Molecular and cellular MOA of a new compound and its associated pharmacokinetics, pharmacodynamics, toxicology and bioavailability properties (Fig. 4)161,162,163,164.

B. AI in clinical trials and real-world practice
Identify biomarkers and patient characteristics that influence drug responses, enabling more efficient and informative trial designs. By optimizing parameters like patient selection, treatment regimens and outcome measurements, ...accelerate the translation of candidate drugs into clinical practice. Real-world data also offer a rich source of information from which AI applications can predict adverse events, drug–drug interactions and other outcomes.
1. Biomarker discovery
... AI further excels in identifying prognostic biomarkers crucial for predicting disease progression and patient survival, thus enabling targeted and personalized treatments.
For example, CD8+ T cell morphology in blood samples -> deep learning models -> identify proteomic biomarkers to accurately169 predict liver disease outcomes.
Prognostic biomarkers for various cancers -> risk scores for survival, recurrence and metastasis. Notably, survival analysis models using graph neural networks outperform existing models, effectively distinguishing risk groups beyond traditional clinical grading and staging170,171
In drug development, predictive biomarkers -> selecting patient populations most likely to benefit from treatments.
The complexity of biological systems necessitates the integration of multiple types of biological data, including protein–protein interactions, into AI models for more comprehensive predictions184.
Future directions for larger datasets
- The integration of datasets from multiple sources shows
- Digital biomarkers from wearable sensors
- Identifying multimodal biomarkers through molecular diagnostics, radiomics and histopathological imaging
- Swarm learning191 and automated dataset processing pipelines192
Challenges
- Heterogeneity (Cellular and tissue-level heterogeneity, diversity of tumor ecosystems, etc...)
- Interpretability T
- Trust
2. Predicting pharmacometrics properties
- Machine learning-based analysis of 442 small-molecule kinases and 2,145 adverse events enabled the discovery of novel kinase–adverse event pairs, enabling risk mitigation and the development of safer small-molecule kinase inhibitors205.
- The multi-omics variational autoencoders (MOVE) framework integrates multi-omics data to reveal drug interactions—such as the link between metformin and the gut microbiota—and compares drug responses across various omics modalities206. PharmBERT, a domain-specific language model, enhances drug safety by extracting crucial pharmacokinetics information from prescription labels207, helping to identify adverse reactions and drug interactions.
- Furthermore, AI can analyze patients’ genetic information, physiological characteristics and past treatment responses to provide personalized dosage adjustment recommendations for doctors, thereby optimizing treatment outcomes208.
3. Drug repurposing
- For instance, AI has accelerated the repurposing of drugs for coronavirus disease 2019 (ref. 209), highlighting the value of AI in finding brand new applications for existing medications.
- AI can also simulate clinical trials using real-world data (including EHRs and insurance claims) to facilitate drug repurposing. As an example of this approach, a deep learning recurrent neural network used causal inference and deep learning to analyze medical claims databases, effectively identifying potential drug candidates. Applied to a cohort of millions with coronary artery disease39.
- Application of deep neural networks to omics data, to classify drugs into therapeutic categories based on the transcriptional perturbations that they induce in vitro210. One study leveraged perturbation samples from the LINCS Project (https://lincsproject.org/) and 12 therapeutic categories derived from MeSH, resulting in high classification accuracy—especially with pathway-level data across diverse biological systems and conditions—offering potential for drug repositioning210.
- Feature attribution techniques, combined with ensembles of interpretable machine learning models, enhance the identification of gene expression signatures associated with synergistic drug responses. This strategy has been shown to improve feature interpretability and support the selection of optimal anticancer drug combinations informed by molecular insights211.
- Furthermore, AI-based high-content screening could also be applied in drug repurposing (Fig. 4). A deep learning model, MitoReID, was developed to identify MOAs through mitochondrial phenotyping.
Challenges
issues with data quality, model interpretability, generalizability, validation costs, regulatory hurdles, integration with existing pipelines and high computational demands.
4. Improving trial efficiency and predicting outcomes
Clinical trials are often expensive, time consuming and inefficient, with the majority facing delays in registration or struggling to find sufficient volunteers. AI has the potential to optimize trial design, streamline recruitment and predict patient responses, improving trial efficiency and success rates while reducing costs and timelines.
- An advanced pipeline has been created that integrates multimodal datasets, generates molecular leads using AI, ranks them by efficacy and safety, and uses deep reinforcement learning to create patentable analogs for testing212. It also predicts phase I/II clinical trial outcomes by estimating side effects and pathway activation, improving prediction accuracy and identifying potential risks in drug portfolios.
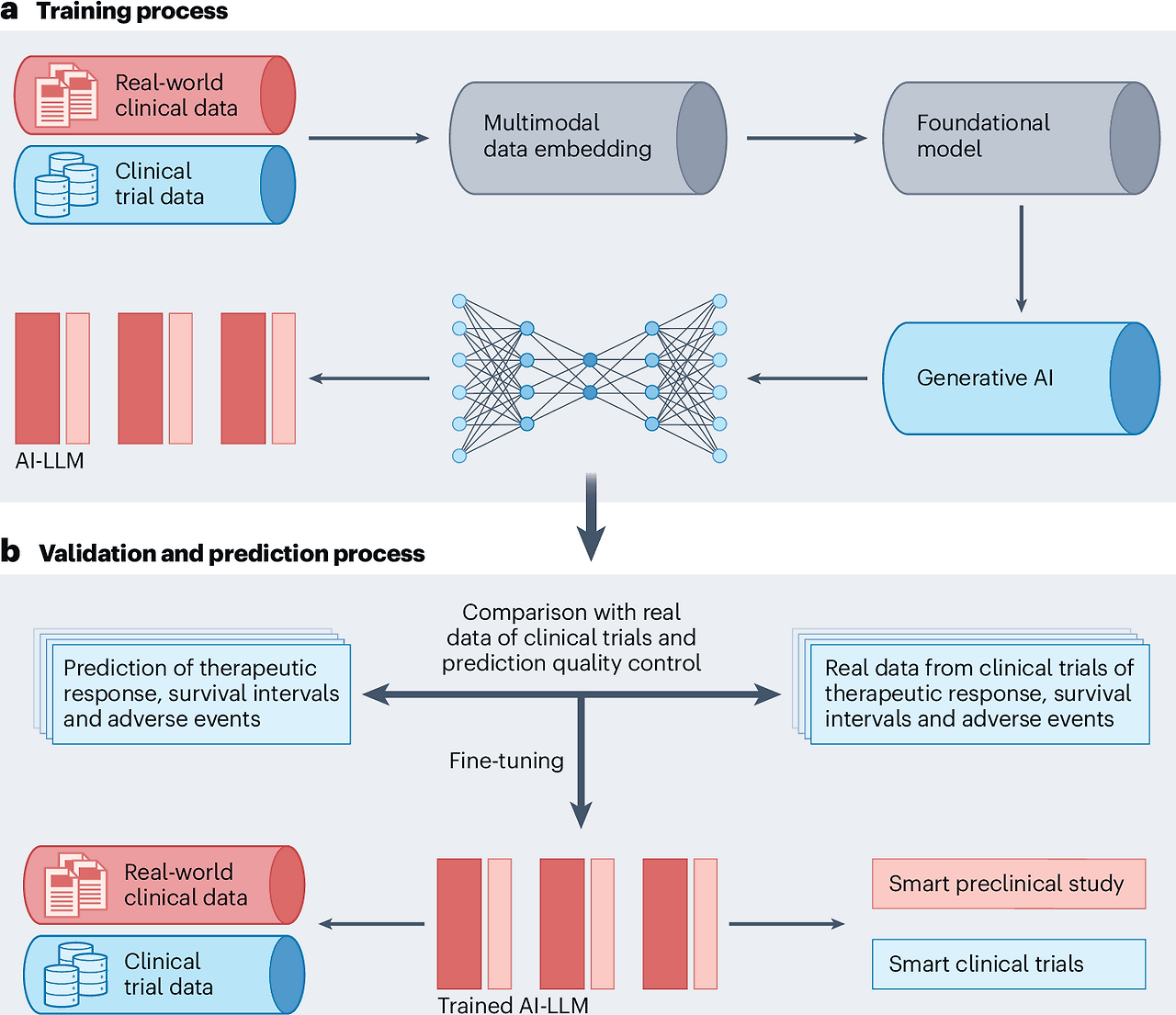
- In real-world studies, AI can analyze data from EHRs, insurance claims and wearable devices to assess drug effectiveness and safety (Fig. 5). For example,Trial Pathfinder tool
- The challenge of finding suitable patients who meet inclusion criteria can be mitigated by using Digital Twins, as explored by Unlearn.ai. This technology creates virtual replicas of participants, allowing them to serve as the control group, thereby increasing the number of participants in the experimental group and improving trial efficiency.
- Beyond the clinical trial stage of drug development, AI can also analyze post-market surveillance data to support the safety, efficacy and quality of drugs.
C. Challenges
Despite advancements, no AI-developed drugs have yet progressed beyond phase II trials, highlighting the complex nature of drug development.
- A key challenge is the lack of high-quality training data
- A key challenge in drug design is balancing multiple objectives for success. Current research often focuses too much on the chemical space, neglecting other key factors (such as druggability and synthesizability). While multi-objective design methods are improving220,221, developing effective scoring functions (for example, for affinity prediction and bioactivity) remains complex and requires considerable experimentation. The absence of standardized evaluation processes further complicates model assessment222,223, especially when conflicting objectives arise, such as maximizing similarity to known bioactive molecules while achieving structural novelty. Although benchmarking platforms like MOSES224 and Guacamol225 exist, a consensus on best practices has not yet been reached.
- Appropriate molecular representation is key in generative models.
- AI faces challenges with so-called ‘undruggable’ targets that lack suitable binding sites
- Finally, technical challenges with algorithms and computing power limit AI’s use in drug development. Many AI algorithms used in drug development were designed for other fields and may not be fully suitable.
D. Future directions
- First, developing new strategies to address the data scarcity issue in AI-enabled drug development should be the top priority.
- Current methods typically focus on single data types, thereby missing complex interrelations between various biological systems. Establishing effective multimodal fusion approaches
- Many current AI models are purely data driven, limiting their effectiveness in drug development due to the relative lack of sufficiently high-quality data. Because our life systems all adhere to the principles of physics (also called the First Principle), drugs also follow the constraints of physical laws without exception. Incorporating physical laws into existing data-driven AI algorithms is one future research direction that could help reduce data dependency and improve both the accuracy and generalizability of these models.
- AI, especially LLMs, can ensure compliance with drug regulations by analyzing extensive documentation and keeping up with the latest requirements./ Interpretable AI models is essential for building trust



